Leber病又称leber遗传性视神经病变,或称家族性视神经病变。由德国眼科医生leber于1871年首先报道,故又称leber病。是引起儿童盲的遗传性疾病之一。
病因:该病遗传方式长期存在争论,既往均认为属于性隐性遗传。临床上可有多种遗传方式,但均不完全符合孟德尔遗传定律。由于在受精时镜子只有头部进入卵细胞,细胞质不进入受精卵而传给下一代,至今尚未发现男性患者可将该病传给后代,均是通过女性垂直传递,提示该病与母亲的细胞质遗传有关,是一种典型的母系遗传性疾病。一般男性较女性发病为多,西方患病率男女比例约为9:1,我国男女比例约为6:4与日本相似,黄种人发病有增多现象。发病多在(18-23岁),我国有趋早发病倾向。
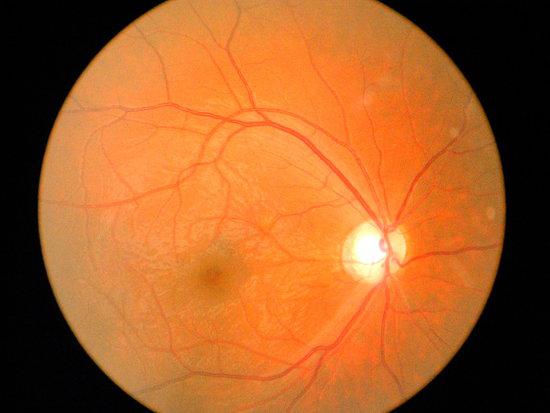
临床表现:双眼常同时或先后发病,多呈急性、亚急性、无痛性发作,其后呈慢性逐渐发展。临床一班可分为临床前期、急性期、亚急性和慢性萎缩期。临床特征呈无痛性视神经病变,急性期视力可几句下降至眼前指数。视盘充血,视盘周围毛细血管扩张性微血管改变、视盘周围视网膜神经纤维层肿胀和视盘无渗漏三联征,慢性期则视盘色淡或苍白。视力大多数在0.1或0.1以下,很少有全盲者。视野中心暗点及旁中心暗点为最多见。有学者认为瞳孔对光反射正常为其特征,色觉障碍为后天获得性,以红绿色盲多见,病情好转,色觉障碍也随之好转,通过色觉检查可预测其发病可能性。FFA在急性期视盘呈强荧光,血管高度扩张,视盘黄斑束毛细血管充盈,延缓缺损等,无渗漏为典型特征,亦可作为早期检测手段。
诊断:根据病史及体征诊断本病并不难,特别有家族发病史者,但与临床上常见的机型视盘炎、球后视神经炎等鉴别并无特殊办法,无家族史者行血液检测mtDNA有无位点突变,正常人及其他视神经病变者无此突变,临床检测有实用诊断价值。
治疗:尚无突破性治疗。有报道称艾地苯醌可激活脑线粒体呼吸链活性,改善能量代谢,是ATP增加,抑制脑内线粒体生成时的过氧化胶质,对该病有一定的效果。该病为母系遗传性疾病,所以遗传咨询十分重要,如为男性患者,其后代不发病,如为女性患者,其子女中男性发病率约为占50%,女性发病率较低,约占20%,但可为携带者。因此,对女性患者或女性携带者更应常规性血液mtDNA检测,明确诊断,以利于优生优育。



 京公网安备 11010802036734号
京公网安备 11010802036734号